
英语原文共 18 页,剩余内容已隐藏,支付完成后下载完整资料
点击化学:
来自几个良好反应的多种化学功能
Hartmuth C. Kolb,MG Finn和K. Barry Sharpless *
摘要:对大自然最喜欢的分子的考察揭示了碳-碳键形成碳-杂原子键的明显偏好,考虑到二氧化碳是自然界的起始材料并且大多数反应是在水中进行的,这一点并不令人惊讶。核酸,蛋白质和多糖是通过碳 - 杂原子键缝合在一起的小亚基的缩合聚合物。即使是35个左右的积木。除了三种芳香族氨基酸之外,这些关键分子各自最多含有六个连续的C-C键。从自然界的方法出发,我们在这里讨论一系列强大,高可靠性和选择性反应的发展,以便通过杂原子链接(CXC)快速合成有用的新化合物和组合文库,我们称之为“点击化学”。点击化学是一次通过一些几乎完美的“弹簧加载”反应来定义,启用和约束。描述获得点击化学状态过程的严格标准,以及使用这种简单但强大的综合策略轻松制作的分子框架示例。
关键词:组合化学,药物研究,合成设计,水化学
1.简介:超越羰基化学的范式
地球上的生命需要在含水环境中建造碳 - 碳键。羰基(羟醛)化学是自然界C-C键形成的主要引擎。在水环境中不仅必需的碳亲电体(羰基化合物)和亲核试剂可以共存,而且水为反应物之间的质子穿梭提供了完美的环境,这对于可逆的羰基化学来说是必需的。
自然界以CO2作为碳源和基于一些较好羰基化学的反应主题,实现了惊人的结构和功能多样性。羰基化学可以用于制造约35个简单构建块的适度收集,然后将其组装成生物聚合物。酶促聚合物与三磷酸腺苷提供的能量增量一起做为选择性催化剂可以阻止基于羰基化学的合成物质崩解混乱。由于许多生物合成途径在每一步都需要独特的酶,因此酶控制策略需要大量的时间和资源用于催化剂开发。由于拥有几十亿年的时间和一个可供使用的星球,自然届既有时间和资源可供选择,但作为人类时间尺度上的化学家,我们并不能这样做。
尽管如此,基于羰基的反应一直深深地吸引着有机化学的学生和从业者。 我们的观点是,有机合成模仿自然界的羰基化学反应并不适合于快速发现具有所需性质的新分子。
许多形成“新”碳 - 碳键的转变只需要被赋予适度的热力学驱动力。特别是,平衡“羟醛”反应通常在低于3kcal/mol下是有利的[1]。为了使这些过程在实验室完成,必须提供额外的“推动”,通常应用Le Chatelier原理(例如共沸去除水),通过将所需过程与放热共聚反应结合(例如,强“碱” 强“酸”)或通过考虑有利的熵变化(例如分子内环闭合)的同时而没有焓的阻碍作用(如形成紧张的环)。因此,实际上由于失去了一个“当量”的酯,共振稳定性是分子间Claisen缩合的第一个步骤(例如,两个乙酸乙酯分子 →乙基乙酰乙酸乙酯 乙醇)是通过吸收约11kcal/mol的热量进行的,但是下一步beta;-酮酯去质子化转化为其烯醇化物的反应中,在提供等化学计量的醇盐时,该反应是放出大量热驱动反应过程的完成。约30年前,动力学控制的烯醇化学的发展,由更强碱如二异丙基氨基锂提供,为这种反应控制提供了最终形式。令人困惑的复杂性的天然产品几乎都是由本领域的精英从业者以这种方式进行常规合成的,所以它显然是一种强有力的方法,而且在这方面最好的工作是作为一个了解关于影响化学反应因素的新见解的丰富来源。但是,如下所述,我们认为这种方法最终会在令人困扰的方向拉动有机合成,最棘手的问题是全球保护的需要和质子功能团体脱保护的复杂性。
如果有用的属性是我们的目标 - 例如更好的药品 - 那么使用复杂的合成策略只有在它们提供了实现这些属性的最佳方法时才是合理的。具有生物活性的天然产物对其感兴趣的合成有机化学家而言具有很难构建的框架,这是因为存在太多连续的碳 - 碳键。然而,这些并不是唯一可以产生有用生物效应的分子。 天然产物合成的悠久和令人钦佩的历史,在保护负载的羰基动力学化学方案中达到高潮,可能使我们忽略了开发合成策略的可能性,而这种可能性也许能够更快速地发现和生产具有期望的分子量的分子属性。
2.“过程化学”的观点
生命系统产生的分子一直吸引和启发着合成有机化学家。随着我们技能和工具的进步,选择用于合成的化合物变得越来越具有挑战性。所以毫不奇怪,现在最喜欢的目标是发现于迄今为止发现最恼人般复杂的天然物质——由植物产生和微生物用于自卫的各种次级代谢产物。这些非凡的分子可以不消耗费用或努力就可以微量合成[2,3]。
制药工业是天然产物化学的直接衍生工业[4],并没有被难题困住的合成方法[5],和现在被探索的许多化合物作为为药物候选体现出的大量合成挑战所阻碍。虽然它提高了研究团队的技能和友好的敬意,但在决定追求复杂药物目标时,隐含着对探索的结构空间的范围需要接受巨大的限制。当天然产物是模型时,通常需要很长时间才能在给定的系列中合成类似物,以至于最雄心勃勃的探索性努力从客观来看往往也是表面的。在这些情况下,探测结构 - 活性关系(SAR)的过程中会在难以合成的领域中有种不良倾向去发现“最佳”候选,接近“可达性”的外部界限。在竞争对手提出最初的发现后,在试图达到可取得专利结构性区域时,也往往会出现困难的合成。
SAR探测和合成足够的更好的化合物用于药代动力学和毒性分析所需的时间是惊人的,并且为了解释为什么这个阶段的药物研究需要很长时间也需要有很长的路要走。然而,在成功获得复杂的目标结构的过程中,发现化学家和高级管理人员对合成可达性问题几乎没有关注。忽视的一个事实是,任何有供应问题的领导或开发系列都有可能存在不合适的SAR结论和开发决策。碳青霉烯类抗生素硫霉素[6-9]的故事是说明性的:它需要由工业界和学术界的几个研究小组花费六年时间才能开发最终治疗药剂(美罗培南[9],是一种噻吩霉素的衍生物)[10a]。艾滋病蛋白酶抑制剂Crixivan(indinavir)提供了一个更近期的例子——是一种在接近日化规模下非常困难的合成[10b]。
只有最后才考虑过程开发的问题,以及生产规模综合而言往往花费巨大。尽管如此,轰动性药物的前景对综合团队来说是一个强大的动力,使得这项工作几乎总能完成。关键的一点是复杂合成对最终药物而言成本虽然很高,但与通过同样复杂的合成方式对发现/发展阶段的速度和质量有强加作用发的所有“隐藏”成本相比,是微不足道的。换句话说,有机合成的方式对整个药物发现,研究和制造过程都有着广泛的影响。如果更模块化,更快的合成方式证明其有效性,降低制造成本应该是其益处中最小的。前体结构不应该是综合性的“宝贵”,而应该能够轻松地从一个系列跳到另一个系列。就目前而言,当寻求功能时,大多数发现的努力都会受到在结构上投入太多的困扰。
考虑大自然如何合成最重要的分子,即主要代谢物。尽管上述次级代谢产物具有广泛的连续碳 - 碳键网络,并且吸引大部分人工合成有机化学家的注意力,但它是个涉及碳 - 杂原子连接的可逆缩合过程,用于装配多核苷酸,多肽和多糖——生命过程中心的三大分子家族。通过采用从小型砌块制造大型低聚物的策略,自然界也是一个完美的组合化学家[11],能够以少于40个单体实现惊人的多样性。除三种芳香族氨基酸外,这些结构单元至多含有六个连续的C-C键。 因此,大自然是碳-杂项连接的混杂创造者,选择这种方法来编码和表达信息。
大自然创造和控制生物分子多样性的能力在很大程度上取决于她部署的精细选择性催化剂。我们用于管理反应性和选择性的设备要复杂得多,特别是在形成C-C键方面。因此,计划合成的化学家需要在可用原料中构建不存在或潜在的C-C键(例如,CH-CX 碱→C=C),这是一个问题,特别是如果这种合成必须是对于许多底物可靠(如组合搜索或SAR研究)或适用于实际的大规模生产。如果不需要从头C--C结构的方式来统一功能化和/或重新组合起始材料和中间体,问题就不太可能发生。如果需要这样的“新”C-C键,最好使它们在分子内形成[12],但最好还是把困难的C-C键的合成留给自然界[13]。
然而,仍然有很大的发现空间:Guida和同事估计了“合理的”候选药物(不超过30非氢原子和500道尔顿,由H,C,N,O,P, S,F,Cl和Br组成;在水和氧气存在及环境温度下可以稳定)在1062和1063离散分子之间.。在这种结构空间下,我们认为在难以到达的地方搜索所需的功能是没有意义的。 相反,我们在这里介绍合成药物发现的方法,遵循一条规则:所有的搜索都必须限制在易于制造的分子上。我们希望让读者相信,可以很容易地制造出各种各样有趣的分子,并且有机会获得的理想生物活性至少与现在药用化学家喜欢的传统目标结构一样好。
2.1 “点击化学”
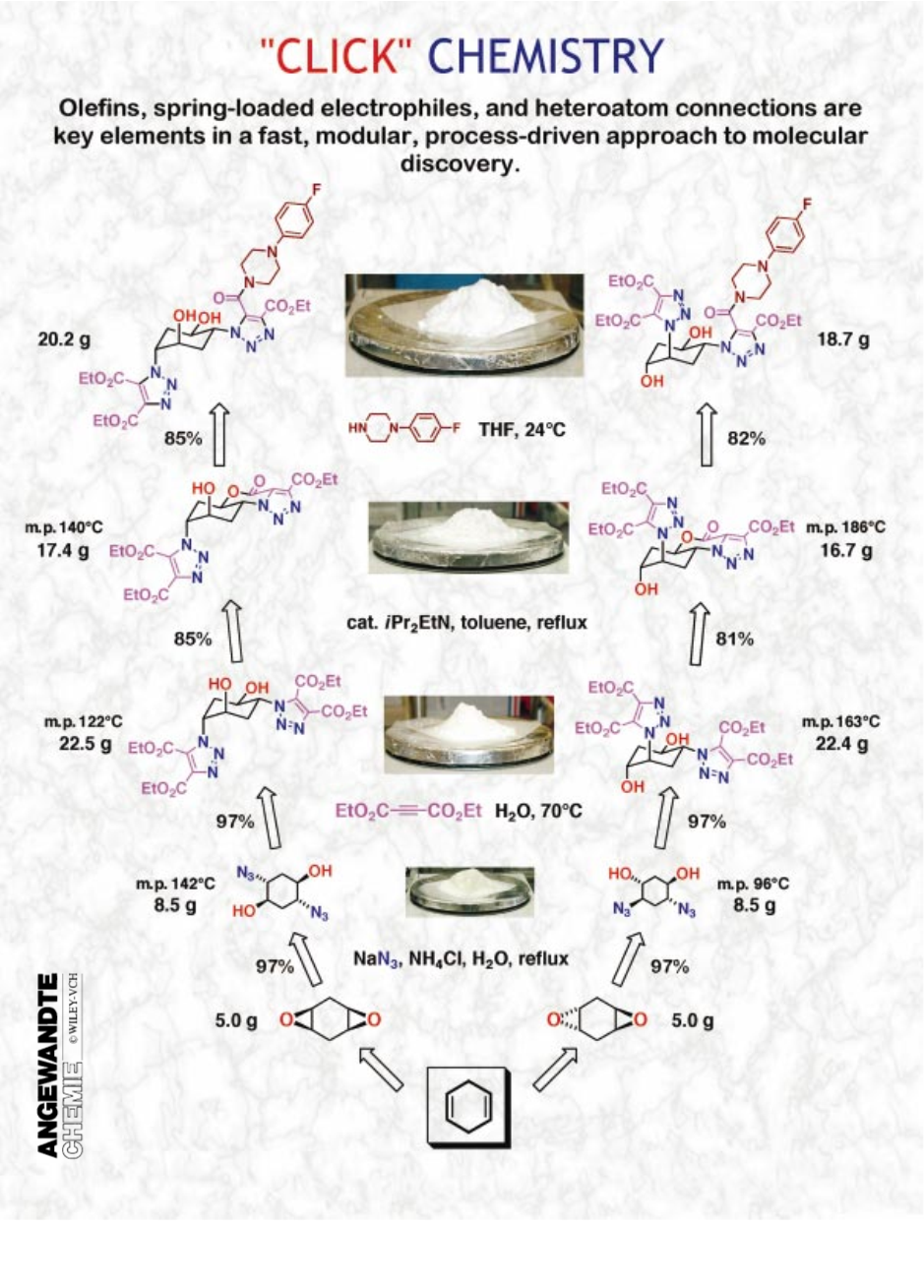
继大自然之后,我们致力于通过将小单元与杂原子链接(C-X-C)结合在一起来生成物质。目标是开发一套功能强大,选择性强,模块化的“模块”,可在小型和大型应用中可靠运行。我们称这种方法的基础是“点击化学”,并且已经定义了一套严格的标准,流程必须满足这些标准才能在这方面有用。反应必须是模块化的,范围广泛,产率很高,仅产生可以通过非色谱方法去除的无害副产物,并且是立体有择的(但不一定是对映选择性的)。所需的工艺特性包括简单的反应条件(理想情况下,该工艺应该对氧气和水不敏感),容易获得的原料和试剂,不使用溶剂或良性溶剂(如水)或容易除去的简单的产品隔离。纯化(如果需要)必须采用非色谱方法,如结晶或蒸馏,产品在生理条件下必须稳定。
重要的是要认识到点击反应达到了它们的要求特征是具有高热力学驱动力,通常大于20 kcal/mol。这样的过程很快就会完成,对于单一产品也往往具有高度的选择性:我们认为这些反应是单弹道“弹簧加载”的。碳 - 杂原子键形成反应组成的最常见的例子,包括以下几类化学转化:
bull;不饱和物种的环加成反应,特别是1,3-偶极环加成反应,还有D-A反应;
bull;亲核取代化学,特别是应变杂环亲电子试剂如环氧化物,氮杂环丙烷,氮杂环丙烷鎓离子和锍鎓离子的开环反应;
bull;“非醛醇缩合”型的羰基化学,例如形成脲,硫脲,芳香族杂环,肟醚,腙和酰胺;
bull;形成碳 - 碳多重键,特别是氧化情况下,例如环氧化,二羟基化,氮丙啶化和卤代亚磺酰基卤化物,还有迈克尔加成的Nu-H反应物
2.2烯烃基有机合成
作为制药工业的对口,考虑石油化工及其衍生材料(纺织品,树脂,塑料等)的世界。石化学家的起始材料是由古代有机体的羰基合成制成的“礼物”;在石油中储存CO2基于光合作用的能量,更重要的是储存无数碳-碳键。然而,几乎完全饱和的天然石油对于大多数有机合成需求是无用的。因此,石油工业是基于对C-C键的操纵,这需要通过“破解”将C-Csigma;键转换为新的C-Cpi;键,并以C-H键为代价通过“改革”创建新的C-Cpi;键。 这些方法的产物是少量反应性单体,然后将其与一批选择性催化剂一起组装成无数有用的材料。因此,基于模块化,高效率的合成策略,石化产品的生产将能源用于“提升”饱和烃成为烯烃似乎是无关重要的。
从一般意义上说,生命化学和石油化学为合成具有不同功能/性质的物质制定了相同的策略:在选择性催化剂的控制下模块化组装专门合成的单体。一个取决于可逆的羰基化学,另一个取决于不可逆的烯烃化学,不过不能掩盖两种方法核心的惊人相似性。我们被剥夺了能够实际模仿大自然的模块化羰基合成方式的能力(见上文),所以石油化学家给我们提供基于烯烃的模块化方法就是我们选择遵循的模型[16]
C-C键作为“货币”单位的概念是理解有机合成中点击化学价值的重中之重。方案1描述了货币如何交换的例子。记录“总C-C键数”的簿记设备通常与可证明的相互转换有关,但这种关联并不是必需的。例如,从正辛烷开始,总C-C键计数保持恒定在七,而不管饱和CC键转化为C=Cpi;键的次数如何,C1-C2-C3-H→C1-H C2=C3。这样的转变是当然是吸热的,但在高温下变得有利,这归功于实质的正熵项。石化行业在大规模实施“蒸汽裂解”(大约850°C的气相法),以提供一系列廉价的烯烃。新创建的C=Cpi;键反应性很高,大约比C-Csigma;键的自由能含量[17]高22 -25kcal/mol;实际上,“裂化”消耗的能量部分被捕获并存储在C=C链中。这在D-A反应和加氢甲酰化等过程中变得明显,它们通过重新组合pi;C=C链获得了连接,同时又不改变总C-C键数。烯烃复分解提供了一种非常容易的方式来重新分配C=C链以供进一步操作。(炔基(计为3个 C-C键),有两个不稳定的pi;键,比烯烃更有利于驱动大量有用的pi;→sigma;碳-碳键重组。)许多这样的过程非常可靠,正是因为没有创建“新”的C-C键; 在反应物的pi;键都是新生的。
方案1. C-C键作为“货币”(1)Delta;H单位为千卡/分子; 加氢甲酰化的给定值仅比较烯烃和醛)。 这些流程中没有形成新的C-C键。
烯烃对合成化学家而言是可用的起始原料中最具吸引力的,可以大量且容易地获得,特别是如果其中包含天然丰富的萜烯。最简单的例子是方案2中显示的一小部分C1-C8块。已经在石化工业中大规模应用,它们是终极重量为90%所有有用的人造有机材料的初始材料[18]。由丁二烯的Wilke环聚异构化合成的环辛二烯和环十二碳三烯是进一步扩大我们可用的适当成本的核心烯烃结构的完美实例[19]丁二烯的pi;键被催化剂毫不费力地重新组合,在每个丁二烯单体后面进行一个烯键连接以进行“装饰”步骤以
全文共45953字,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[13323],资料为PDF文档或Word文档,PDF文档可免费转换为Word
以上是毕业论文外文翻译,课题毕业论文、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。
您可能感兴趣的文章
- 通过对奥美拉唑合成反应的监测和定量反应的在线拉曼光谱和表征组件外文翻译资料
- 无金属碳基催化剂的研究进展外文翻译资料
- 钼酸钙/碳三维复合材料可控设计合成的研究外文翻译资料
- 生物催化选择性合成功能化喹唑啉酮衍生物外文翻译资料
- 三元V Zr Al ON氧氮化物-3-甲基吡啶氨氧化的高效催化剂外文翻译资料
- 综述纳米零价铁(nZVI)的合成,特性和在环境修复中的应用外文翻译资料
- 自消毒PVC表面使用点击化学设计外文翻译资料
- 微波辅助直接合成4H-1,2,4-苯并噻二嗪1,1-二氧化物衍生品外文翻译资料
- 微波辅助下直接合成1,1-二氧代-4H-1,2,4-苯并噻二嗪类衍生物外文翻译资料
- 压力选择在变压精馏中的重要性外文翻译资料

