
英语原文共 6 页,剩余内容已隐藏,支付完成后下载完整资料
应变引起的具有较高反常霍尔电导率的二维Cr2Ge2Se6室温铁磁半导体
Xue-Juan Dong,1 Jing-Yang You,1 Bo Gu,2,3* and Gang Su1,2,3dagger;
1中国科学院大学物理学院, 邮编:100049 中国北京
2Kavli 理论科学研究所和中国科学院大学CAS拓扑量子计算研究中心,
邮编:100190 中国北京
3怀柔国家综合科学中心物理科学实验室,邮编:101400 中国北京
(收于2019年1月16日; 修正稿收于2019年5月22日; 出版于2019年7月11日 )
通过密度泛函理论计算,我们预测到一种稳定的二维(2D)铁磁半导体Cr2Ge2Se6,在该半导体中,居里温度TC可以通过施加几个百分点的应变提高到室温以上。而且二维Cr2Ge2Se6和二维Cr2Ge2Te6的反常霍尔电导率预计与铁磁性金属铁和镍的反常霍尔电导率相当,并且比稀磁半导体镓(锰、砷)的反常霍尔电导率大一个数量级。基于超交换相互作用,我们可以通过减小Cr的3d轨道与Se的4p轨道间的能量差来解释由应力引起的二维Cr2Ge2Se6的TC的增加。我们的发现强调了通过应变获得室温铁磁半导体的微观机制。
1.介绍
磁性和半导体的结合使得磁性半导体得以发展,这是一种基于电荷和自旋自由度在电子器件中实现自旋电子应用的有前途的方法[1,2]。其中研究最广泛的磁性半导体(Ga, Mn) As所具有的最高居里温度TC为200 K [3],这显然还是远低于室温的。而现在我们迫切的需求可在室温下运用的铁磁半导体。而最近在二维范德瓦尔斯材料磁性研究中取得的新的进展为磁性半导体的研究提供了新的平台[4]。我们在实验中可以观察到单层CrI3上具有外层磁化的铁磁性,其TC为45K[5]。在双层二维Cr2Ge2Te6的实验中可获得为TC为28K的海森堡铁磁态[6],并且实验证明相应的堆积体是层状铁磁体,其沿c轴自旋,Tc = 61 K [7]。根据最近的实验,在拓扑绝缘子(Bi, Sb)2Te3上的Cr2Ge2Te6 6-nm薄片上有一种具有面外磁各向异性的较强剩余磁化的TC为80K[8]。有关于Kitaev相互作用的单层CrI3和Cr2Ge2Te6磁化结构最近也受到广泛讨论[9]。同样光致发光和磁光效应也受到研究人员的关注[10,11]。在对单层的VSe2 [12]和MnSe2 [13]中的实验里同样也发现了具有较高TC的铁磁性物质。尽管通过实验实现的单层铁磁材料相对较少,但通过第一性原理计算预测有希望的候选材料可以为实验提供可靠的参考[14,15]。计算机研究[16]和高通计算[17]两个领域同样也在研究潜在的2D铁磁性物质。
在最近发现的这些2D物质中我们已使用了多种方法来控制它们的磁极态。比如通过调整触发电压和在实验获得的双层CrI3[18]中变换反铁磁(AFM)态和铁磁态。通过使用触发电压,我们在二维Fe3GeTe2的实验中观察到了铁磁态的产生[19]。在非磁性二维过渡金属双卤材料中,应力被用来调整光学和电学性能[20]。从中发现应力可以影响单层Cr2Ge2Te6 [21]和CrX3 (X =Cl, Br, I) [22]的铁磁性。其中电场效应也被用于具有磁性的多层Cr2Ge2Te6 [23]的实验研究,最近这些2D物质的异质结构也开始成为研究人员研究的对象[24–26]。
在这篇论文中,我们通过密度泛函理论(DFT)的计算预测到一种2D铁磁性半导体Cr2Ge2Se6。我们发现通过施加3%的应力后,Cr2Ge2Se6的TC可达到144K,施加5%的应力则可达到421K。另一方面Cr2Ge2Te6半导体的TC在我们的计算中大致为30K,这与我们最近的实验中获得的28K的值相近[6]。而且Cr2Ge2Se6和Cr2Ge2Te6中的反常霍尔电导率与铁磁性金属Fe 和 Ni [27–29]的反常霍尔电导率的值相接近,并比稀释磁半导体Ga(Mn, As)的反常霍尔电导率大一个数量级[30,31]。我们还发现基于超交换机制,应力可以减小Cr的3d轨道与Se的4p轨道间的能量差,而且可以增强铁磁耦合。我们的发现其强调了使用应力来获得室温下的铁磁半导体的微观机制。
2.计算方法
DFT计算是通过维也纳从头计算模拟包进行的[32]。我们用投影增广波法进行自旋极化计算,并采用Perdew- Burke-Ernzerhof交换相关函数中的广义梯度近似的方法。自旋轨道耦合也包括在计算中,我们用Mohnkhorst-Pack的方法得到了9times;9times;1 k网格的总能量。在能量收敛小于10-6 eV 和力的收敛小于0.01 eV/Aring;的情况下,我们对晶格常数和原子坐标进行了优化,其中利用值为20 Aring;的真空层对二维系统进行了建模,而声子的计算采用了密度泛函微扰理论和PHONOPY[33]。超胞的尺寸为3times;3times;1,其中位移取0.01 Aring;。
对于Cr离子的现场库伦相互作用U,三维过渡金属[34]绝缘子通常取值为3–5 eV,而我们在二维Cr2Ge2Te6[6]的DFT计算中则采用了较小的值(U lt; 2 eV)。在我们对大多Cr2Ge2Te6和Cr2Ge2Se6的后续计算中,我们将U固定在4 eV,并在以后的研究中探讨了取不同U值时的影响。在DFT结果的基础上,我们通过基于二维Ising模型的蒙特卡罗模拟[21,22]计算了居里温度,其中采用了大小为60times;60的超级单元,并对每个温度进行了多达105个步骤以达到平衡,之后用WANNIER90 [35]和Wannier Tools [36]计算了反常霍尔电导率。
3.晶体的稳定性
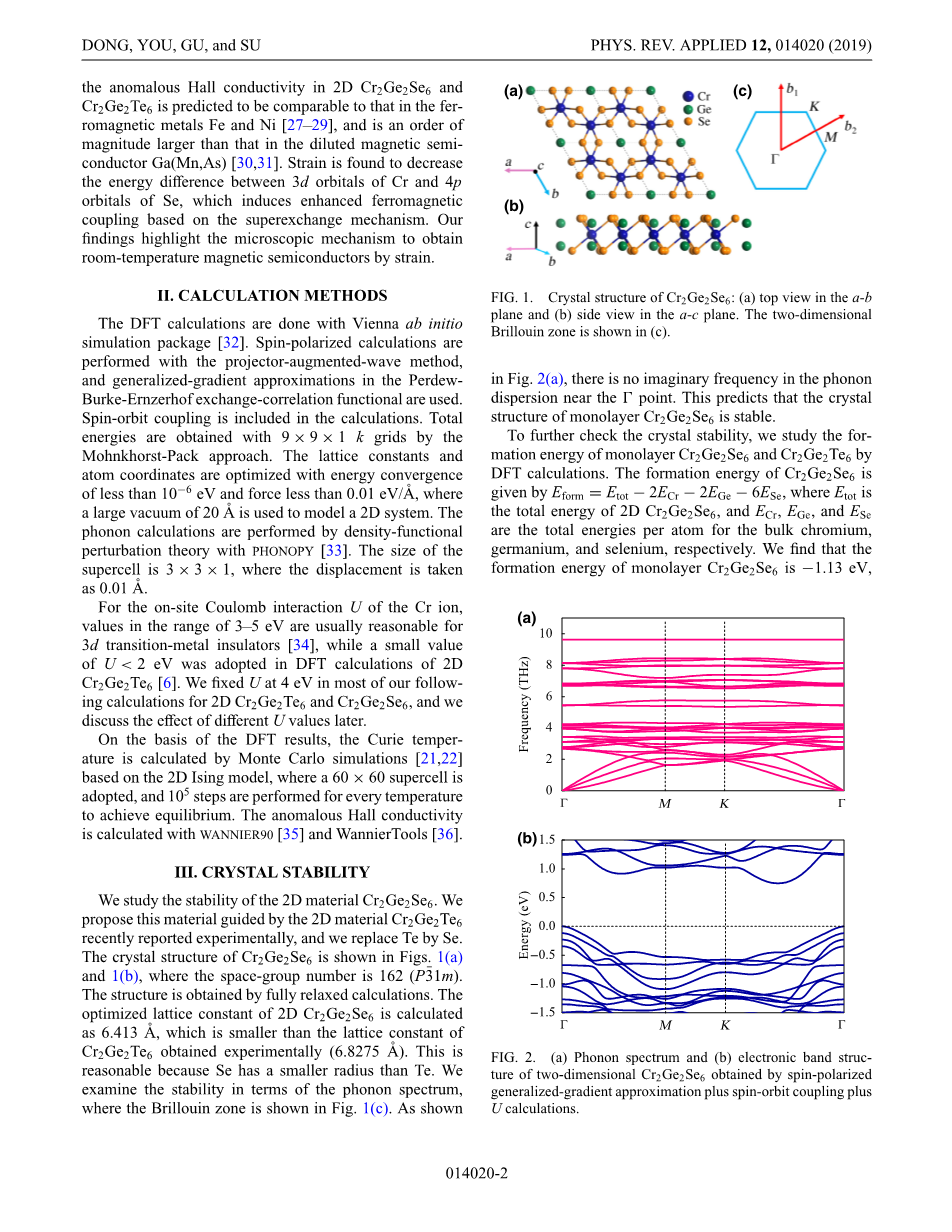
我们研究了二维材料Cr2Ge2Se6的稳定性。鉴于近期实验的结果我们提出了将Cr2Ge2Te6中的Se取代了Te得到。图1(a)显示了Cr2Ge2Se6的晶体结构,图1(b)中的空间群号为162 (P3macr;1m),该结构是通过充分简化后的计算得到的。通过计算,二维Cr2Ge2Se6的优化晶格常数为6.413 Aring;,其值小于实验得到的Cr2Ge2Te6的晶格常数(6.8275 Aring;),因为Se的半径比Te的小,所以这样的结果是合理的。之后我们用声子谱的方法研究了其稳定性,其中布里渊区如图1(c)所示。而如图2(a)所示,在点T附近的声子色散中没有虚频率,这预测了单层的Cr2Ge2Se6的晶体结构是稳定的。
(a)
(c)
(b)
图1. Cr2Ge2Se6的晶体结构: (a) a-b平面的顶端视图,(b) a-c平面的侧视图,(c)二维布里渊区。
为了进一步验证晶体的稳定性,我们通过DFT计算研究了单分子层Cr2Ge2Se6和Cr2Ge2Te6的形成能,Cr2Ge2Se6的形成能公式为Eform = Etotminus;2ECrminus;2EGeminus;6ESe,其中Etot是Cr2Ge2Se6的总能量, 而ECr, EGe, ESe分别是铬、锗和硒的每个原子的总能量,我们发现单层Cr2Ge2Se6的形成能为minus;1.13 eV,这要比单层的Cr2Ge2Te6所具有的0.92 eV还要低,这表明二维的Cr2Ge2Se6的结构应该比二维的Cr2Ge2Te6更稳定(Cr2Ge2Te6形成能来
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[245468],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 复杂热电材料外文翻译资料
- 以自蔓延高温烧结方法制备热电化合物以及燃烧合成的新标准外文翻译资料
- 氮掺杂分级多孔碳作为氧还原反应的高效电化学催化剂的研究外文翻译资料
- 孪晶诱导塑性高嫡合金的设计外文翻译资料
- 含铌先进Fe-Cr-Ni型奥氏体耐热钢富铜相的析出强化在超临界电厂的应用外文翻译资料
- 不同温度下直接能量沉积层状工具钢的弯曲强度外文翻译资料
- BiFeO3的光伏效应外文翻译资料
- 通过氢稳定的MgaPt研究核壳纳米结构Mg@Pt中快速“氢泵”的可视化外文翻译资料
- 一种铱核心环金属有机配体显著地提高了有机太阳能电池 的光伏性能外文翻译资料
- 钠离子电池的高性能阳极材料:三组分共组装法制备层次多孔碳外文翻译资料

