
英语原文共 5 页,剩余内容已隐藏,支付完成后下载完整资料
3d过渡金属掺杂MoSe2单层的电磁学性能
Yi Tian, Zhipeng Zhu, Zhizhong Ge, An Sun, Quan Zhang, Songlei Huang, Hongping Li*, Jian Meng
江苏大学材料科学与工程学院新材料研究所,镇江,212013,中国
中国科学院长春应用化学研究所稀土资源利用国家重点实验室,吉林长春,130022,中国
摘要
化学掺杂是调节材料性能的最有效方法之一。本文对掺杂3d过渡金属Mn、Fe、Co和Ni对单层MoSe2的原子结构和电子性质的影响进行了系统的研究。结构分析表明,尽管晶格略微扭曲,但所有掺杂体系几乎保持了MoSe2原来的结构类型。形成能表明富Se条件下的形成能比富Mo条件下更有利,而在这两种条件下,Mn的掺入都是很有利的。通过在能带中引入平坦的杂质带,电子输运特性得到了增强。在掺Fe的情况下实现了半金属的转变。掺杂Mn、Fe、Co、Ni等杂质时单层MoSe2总磁矩分别为1.074mu;B、1.963mu;B、2.760mu;B、1.765mu;B,磁性特征明显。我们的研究结果表明,过渡金属掺杂是调制单层MoSe2性能的有效策略,以MoSe2的技术应用为目标。
关键词:MoSe2单层,过渡金属掺杂,电磁学性能,第一性原理计算
1.引言
层状过渡金属二硫化物(LTMDs)是一种类石墨烯材料,由于其具有独特的性能,如突出的柔韧性、适度的载流子迁移率、独特的电子和光学性能而被广泛研究,使其成为器件应用的通用候选材料。在结构上,LTMDs的化学式为MX2 (M:过渡金属,X:硫代物),它由两个紧密排列的X层由一个M原子层隔开,形成了在平面上具有强共价键的共边MX6八面体。然而,相邻的X-M-X层通过弱范德华力(vdW)相互耦合,为剥落成超薄层提供了可行性。在过去的几年里,不同的二维MX2 (M=Mo, W;X=S, Se, Te)通过物理或化学方法制备了三层原子层厚度的薄板。结果,它出现了一些独特的性质,这些特性不同于它的体积对应物,例如直接带隙跃迁[1,2],选择性光耦合[3,4],非线性光响应[5],这为它投入预期应用开辟了新的途径。
近年来,许多可行的方法被用于调节单层LTMDs的物理特性以实现其潜在的应用,如缺陷[6]、杂质掺杂[7]、外部应变[8]和化学吸附[9]。在这些方法中,掺杂被认为是调节原子结构和电子功能的有效方法。在以往研究的基础上,过渡金属掺杂已成功地用于调整其性能。例如,通过加入Nb[10],在MoS2中实现了p型导电。单个Co原子掺杂将半导体单层MoS2转化为半金属[11]。在单层MoS2中引入一系列金属V、Mn、Fe、Co、Ni原子[7],并通过过渡金属 d带填充[12]增强其磁性。特别是在单层MoS2中,由于Cu与相邻的S原子[7]有很强的杂化作用,非磁性Cu掺杂可以获得较大的间隙磁矩。另一方面,掺杂到MoS2单层中的非金属元素,如H、B、N、Cl、F等,也可以诱导出好的电子特性和不可忽略的自旋极化[13-15]。因此,掺杂是优化单层LTMDs电子结构和磁性能的有效途径,拓宽了其在新器件中的应用前景。
MoSe2单层作为LTMDs中的一个典型代表,因其具有直接带隙[16]、强光致发光[17]和良好的物理特性[18]而引起了广泛的关注。同样,它的电子和磁性也可以通过化学掺杂来控制。事实上,W掺杂单层MoSe2可以通过增强光致发光强度[19],将主导传导从n型切换到p型。在Cu掺杂的二维MoSe2单层中,根据不同的掺杂构型[20],可以获得长程铁磁或铁磁耦合。在以往研究的启发下,期望其他过渡金属可以用于调控MoSe2单层的电子结构。在这里,我们采用第一原理计算,揭示了3d过渡金属Mn、Fe、Co和Ni掺杂对MoSe2单层结构、电子和磁性能的影响。我们的结果表明,在富Se条件下,所有掺杂体系在能量上都对金属杂质有利。更重要的是,不仅带结构被3d 过渡金属杂质显著调谐,而且在所有掺杂系统中都引入了明显的磁矩。
2.计算方法
自旋极化密度泛函理论(DFT)计算使用投影增强波(PAW)[21]作为实现VASP[22]的工具。在Perdew-Burke-Ernzerhof (PBE)[23]形式的广义梯度近似(GGA)下描述了电子交换和相关效应。平面波展开的截断能设定为400 eV,布里渊区积分采用Monhkorst-Pack方案和5times; 5times;1 k点网格[24]进行。为了避免相邻晶胞之间的相互作用,引入了15 Aring;真空间距,从而使构建单层MoSe2成为可能。价电子构型为Mo:4d55s1,Se: 4s24p4,Mn: 3d54s2, Fe: 3d64s2, Co: 3d74s2和Ni: 3d84s2。两离子阶间自洽过程的收敛判据设为1.0 times;10-5ev/atom,每个原子的Hellmann-Feynman力小于0.01 eV/ Aring;。
3.结果和讨论
3.1优化几何结构
单层MoSe2属于具有P6m2空间群的六方晶体结构,其中Mo原子与6个最近的Se原子处于三方棱柱配位。采用4times;4times;1 超晶胞模拟了过渡金属掺杂体系(如图1所示),其中一个Mo原子被一个过渡金属原子取代,掺杂浓度为6.25%。几何结构和原子位置完全松弛。
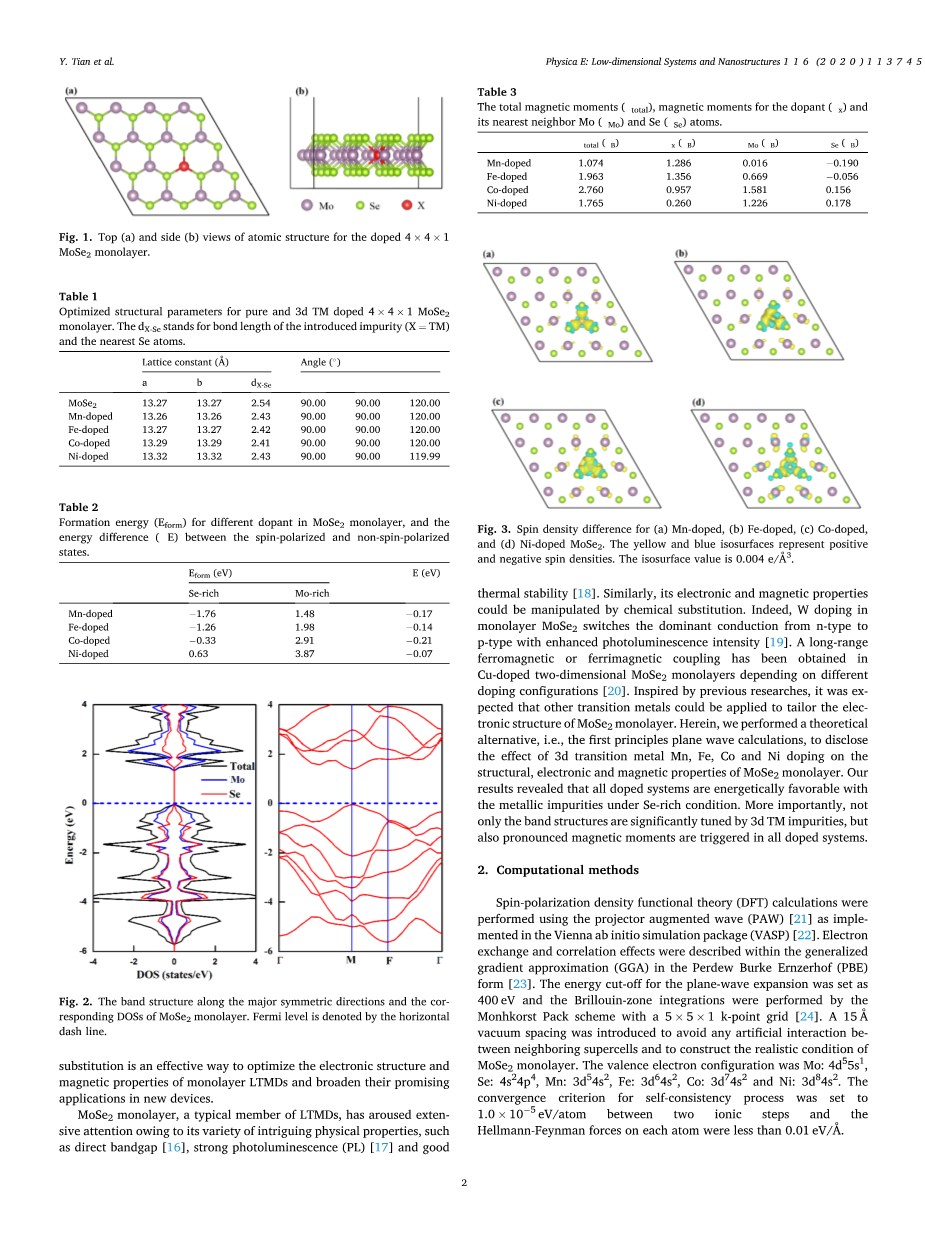
图1 掺杂的4times;4times;1MoSe2单层原子结构的俯视图(a)和侧视图(b)。
优化结果如表1所示,给出了原始MoSe2单层超晶胞的松弛晶格参数进行了比较。显然,所有掺杂体系几乎都保持了MoSe2单分子层的原始结构类型,但由于3d过渡金属的掺杂,晶格有轻微的畸变。这是意料之中的,因为Mn、Fe、Co、Ni和Mo原子的离子半径更近,这表明3d 过渡金属杂质的掺入应该会导致最小的结构畸变。此外,X- Se (dX-Se, X表示过渡金属掺杂剂)的松弛键长比MoSe2单层中最优的Mo-Se键(2.54 Aring;)短,表明它们之间存在较强的键相互作用。
表1.本征和掺杂3d TM的4times;4times;1MoSe2单层的优化结构参数。dX-Se表示引入的杂质(X=TM)和最近的Se原子的键长
3.2形成能
为了评估掺杂了杂质的MoSe2单分子层的相对稳定性,根据以下表达式计算形成能(Eform):
Eform=E(doped)-E(pure) mu;Mo-mu;X
其中E(doped)和E(pure)分别为掺过渡金属和不掺过渡金属时MoSe2超晶胞单层总能量。mu;Mo和mu;X分别为Mo原子和过渡金属掺杂剂(Mn、Fe、Co和Ni)的化学势。众所周知,形成能在很大程度上取决于实验条件。在本研究中,我们考虑了两个限制的合成条件,即富Mo和富Se情况。对于富Mo的情况,假定mu;Mo是每个Mo原子在其稳定体相中的能量。然而,对于富Se的情况,mu;Mo和mu;Se的关系为:mu;Mo 2mu;Se=mu;(MoSe2)。因此,mu;Mo是由一个Se2分子与一个MoSe2单位的能量差来确定的。
表2列出了在富Mo和富Se条件下,3d过渡金属掺杂MoSe2单分子层的理论形成能。值得注意的是,在富Se条件下,由于形成能量较小,过渡金属杂质掺入MoSe2单层的热力学优先于富Mo条件。此外,Eform随过渡金属原子序数的增加而增加,这与MoS2[25]掺杂的结果相似。Mn掺杂的MoSe2的Eform最小,尤其是富Se生长的MoSe2,最有利于Mo与Mn的置换。
表2 MoSe2单分子层中不同掺杂剂的形成能(Eform)以及自旋极化态与非自旋极化态之间的能量差△E
3.3 MoSe2单层的电子性质
为了阐明3d过渡金属掺杂对基体材料电子性能的影响,我们首先对MoSe2单层进行了电子结构计算。图2给出了沿第一布里渊区高对称方向上的态密度和相应的能带结构。显然,MoSe2单层表现出半导体性质,在费米能级上没有分支。由于价带顶和导带底都位于高对称点M处,从而是直接带隙,这与之前的结果[26]一致。自旋向上和自旋向下的完全对称揭示了MoSe2单分子层的非磁性态。Mo和Se的偏态密度由于它们具有相同和广泛的能量分布而表现出很强的共价杂化。
图2 沿主要对称方向的带结构和相应的MoSe2单层态密度。费米能级用水平虚线表示
3.4 3d 过渡金属掺杂MoSe2单层的电子和磁性能
为了揭示3d 过渡金属掺杂剂在MoSe2单层中诱导的电子特性和磁行为,考虑了自旋极化态和非自旋极化态之间的能量差。从表2可以看出,自旋极化态比相应的非自旋极化态在能量上更有利,这意味着Mn、Fe、Co和Ni掺杂形成了磁阵列。对于Mn、Fe、Co和Ni掺杂的情况,计算得到的总磁矩(如表3所示)分别为每个超晶胞的1.074mu;B、1.963mu;B、2.760mu;B和1.765mu;B。对于Mn掺杂的MoSe2单层,磁矩主要分布在Mn掺杂剂上,而很少的磁矩分布在周围的Mo原子上(0.016mu;B)。最近的Se原子携带磁矩为0.190mu;B,表明Mn和Se原子之间存在反铁磁耦合。在Fe掺杂体系中,Mo的磁矩相对较大,每个相邻Mo原子的磁矩为0.669mu;B。而在Co和Ni掺杂的MoSe2单层中,掺杂剂对总磁矩的贡献相对较小,分别只有34.7%和14.7%。
表3 总磁矩(mu;total),掺杂剂的磁矩(mu;x)和它最近的原子Mo(mu;Mo)和Se(mu;Se)
结果表明,最近的Mo原子和最近的Se原子贡献了总磁矩,并与掺杂剂形成铁磁耦合,这与单Cu原子掺杂的MoSe2单层[20]非常相似。特别地,近端Mo原子比掺杂剂和非金属Se原子具有更大的磁矩。所有掺杂体系三维电荷密度差的等值线图作为掺杂剂与MoSe2单层之间电荷转移的直接证据。如图3所示,电荷密度从掺杂剂扩展到最近的Se和Mo原子,这表明它们之间有很强的共价键。此外,在最近的空间扩展的电荷密度Mo和Se原子中发现了Co和Ni掺杂的MoSe2单层,而他们在Mn相对较少,Fe掺杂下,Mo和Se原子周围有更大的磁矩。
图3 (a) Mn掺杂,(b) Fe掺杂,(c)Co掺杂和(d) Ni掺杂MoSe2的自旋密度差。黄色和蓝色等表面代表正和负自旋密度。等值面值为0.004 e/ Aring;3
图4为过渡金属掺杂MoSe2单层的带结构。显然,在所有掺杂体系中,与MoSe2 超晶胞中的Mo原子相比,过渡金属掺杂物中额外的3d电子刺激了费米能级向更高能量区域移动。通过过渡金属杂质的掺入,在能带隙内引入了平坦的杂质带,表明了过渡金属杂质增强了电子输运特性。此外,所有的能带结构在自旋向上和自旋向下通道之间具有明显的不对称性,表明3d 过渡金属掺杂剂可以诱导自旋极化态。这些结果与上述磁矩分析结果一致。在掺Mn的情况下,其自旋向上和自旋向下通道的半导体带隙分别为0.14 eV和1.28 eV(图4(a))。在Co和Ni掺杂的系统中也得到了类似的情况,其中半导体的禁带宽度分别为0.42 eV、0.16 eV和0.76 eV,自旋向上和自旋向下通道的带隙为0.71 eV(图4 (c)、(d))。同样的,在掺杂Cu的MoSe2单分子层[20]中,还可以产生一个在两种自旋通道中都有明显间隙的磁性半导体。然而,对于Fe掺杂体系(图4(b)),几个自旋向上的带穿过费米能级并产生金属性,而自旋向下的带结构仍然是半导体,带隙为0.92 eV。这是一个典型的半金属行为,其中接近费米能级的100%自旋极化对于自旋电子器件的应用是非常有用的。
图4 (a)Mn掺杂,(b) Fe掺杂,(c)C
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[258498],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 复杂热电材料外文翻译资料
- 以自蔓延高温烧结方法制备热电化合物以及燃烧合成的新标准外文翻译资料
- 氮掺杂分级多孔碳作为氧还原反应的高效电化学催化剂的研究外文翻译资料
- 孪晶诱导塑性高嫡合金的设计外文翻译资料
- 含铌先进Fe-Cr-Ni型奥氏体耐热钢富铜相的析出强化在超临界电厂的应用外文翻译资料
- 不同温度下直接能量沉积层状工具钢的弯曲强度外文翻译资料
- BiFeO3的光伏效应外文翻译资料
- 通过氢稳定的MgaPt研究核壳纳米结构Mg@Pt中快速“氢泵”的可视化外文翻译资料
- 一种铱核心环金属有机配体显著地提高了有机太阳能电池 的光伏性能外文翻译资料
- 钠离子电池的高性能阳极材料:三组分共组装法制备层次多孔碳外文翻译资料

