
英语原文共 6 页
对光化反应生产中混合无机-生物有机体系能量转移的光谱学说明
Nikolay Kornienkoa,1, Kelsey K. Sakimotoa,1, David M. Herlihya , Son C. Nguyena , A. Paul Alivisatosa,b,c, Charles. B. Harrisa , Adam Schwartzbergd , and Peidong Yanga,b,c,2
a加利福尼亚大学伯克利分校化学系,CA 94720;b加利福尼亚大学伯克利分校材料科学与工程系,CA 94720;c伯克利卡夫利能源纳米科学研究所,CA 94720;以及d劳伦斯伯克利国家实验室的分子实验室,CA 94720
本文于2016年8月30日批准并由宾夕法尼亚州立大学Thomas E. Mallouk编辑 (2016年6月28日收到供审查)
摘要:用于太阳能化学生产的无机-生物混合有机体系的兴起促使人们对生物-非生物界面的动力学进行机械研究,以推动下一代体系的发展。一个典型体系,热醋穆尔氏菌-硫化镉(M. thermoacetica-CdS),将一种无机半导体纳米颗粒光收集器与一种能产生乙酸的细菌结合起来可以高效驱动光合作用将CO2转化为醋酸。在这篇文章中,我们揭示了这一独特的电营养行为并提出了一种从CdS到M. thermoacetica的电荷转移机理。瞬态吸收光谱显示光激发的电子转移速率随着氢化酶活性的增加而增加。在与瞬态吸收光谱相同的时间范围内,时间分辨红外光谱显示光谱在1.700–1.900 cmminus;1的光谱范围内变化。在光合作用平均第一个24小时内,该系统光合作用乙酸生成的量子效率随着氢化酶活性的增加和载流子寿命的减少而增加。然而,在光合作用开始的3小时内,速率遵循着一个相反的趋势:具有最低氢化酶活性的细菌光合作用生成乙酸的速度最快。这些结果揭示了一个两条途径的反应机理:一种是高量子效率电荷转移路径,即氢化酶产生氢气作为一个分子中间体,这一途径在长时间范围内(24h)占主导地位,而一种直接的能量转换催化途径对乙酸的生成在短时间范围内(3h)起作用。 这一研究为运用传统的光谱方法来理解更加复杂的生物-非生物混合体系提供了一个具有前景的平台。
关键词:能量转换;光谱学;CO2减少量;生物混合体系;催化作用
太阳能通过自然光合作用形成化学键的转化速率缓慢从而阻碍了我们试图获取全部太阳能量的工作(1)。我们通过利用有大量储存量的碳质能源暂时回避了这一局限性,并促进了工业、农业、城市和人口的快速发展。然而,石油化学品日益减少的情况需要光合作用的回归,一种能够与现代社会发展同步的新型光合作用(2,3)。作为一个进步的标志,无机半导体光收集器的效率普遍大于植物(4)。相比之下,合成催化剂仍然难以复制生物学中复杂的C-C键的形成(5,6)。通过对几种无机-生物混合体系即将无机半导体光收集器和微生物CO2减少量结合起来构成的混合体系的研究,对太阳能至化学品生产的理解已取得了重大进展(7-9)。近期,我们报告了一种自身光敏化的混合细菌M. thermoacetica-CdS,该细菌通过生物作用的CdS纳米粒子进行光合作用将CO2转化为乙酸(10)。
尽管电子有几种途径从电极转移到细菌,但转移机理仍存在争议(11)。生电型微生物燃料电池中电子从细菌到电极阳极转移的光谱研究与细胞色素介导的机制有关(12)。然而,关于电营养生物中阴极电子在半导体-细菌间转移的类似研究并不多见。据推测,电子先转移到膜结合处或细胞外的氢化酶上,进而产生氢气,氢气作为中间体随后进入WLP路径(13-15)。由于很难将以前的技术应用到固体电极平台,所以具体的光谱特征仍然是不确定的。相比之下,我们的模型系统M. thermoacetica-CdS是一种半透明的胶体悬浮液,在其中的光位点上CdS纳米粒子产生光电子,抵消了对不透明电极的需要,因此在揭示电荷转移机理的分子基础时使现有的基于透光率的光谱学成为了可能(图1A)。在此,我们提出了与生物化学活性相关的瞬态吸收光谱(TA)和时间分辨红外光谱(TRIR),以建立无机-生物电荷和能量转移动力学模型。
意义:
利用二氧化碳生产太阳能化工产品有望减少石油化学品的消耗。无机半导体光收集器和微生物催化剂的混合系统提供了一条可行的前进道路。尽管已经描述了许多这样的系统,半导体到细菌的电子传输机制仍然是未知的,限制了改善其性能的合理方法。在这项研究中,我们探究了半导体纳米粒子敏感细菌如何将CO2和光转化为乙酸,这是一种已知的燃料、食品、医药以及聚合物的前体。运用时间分辨光谱法和生物化学分析法,我们得出结论:多种途径促进电子和光能从半导体转移到细菌。这一基础性的研究使得对复杂生物-非生物混合体系的未来的研究、理解和提升成为了可能。
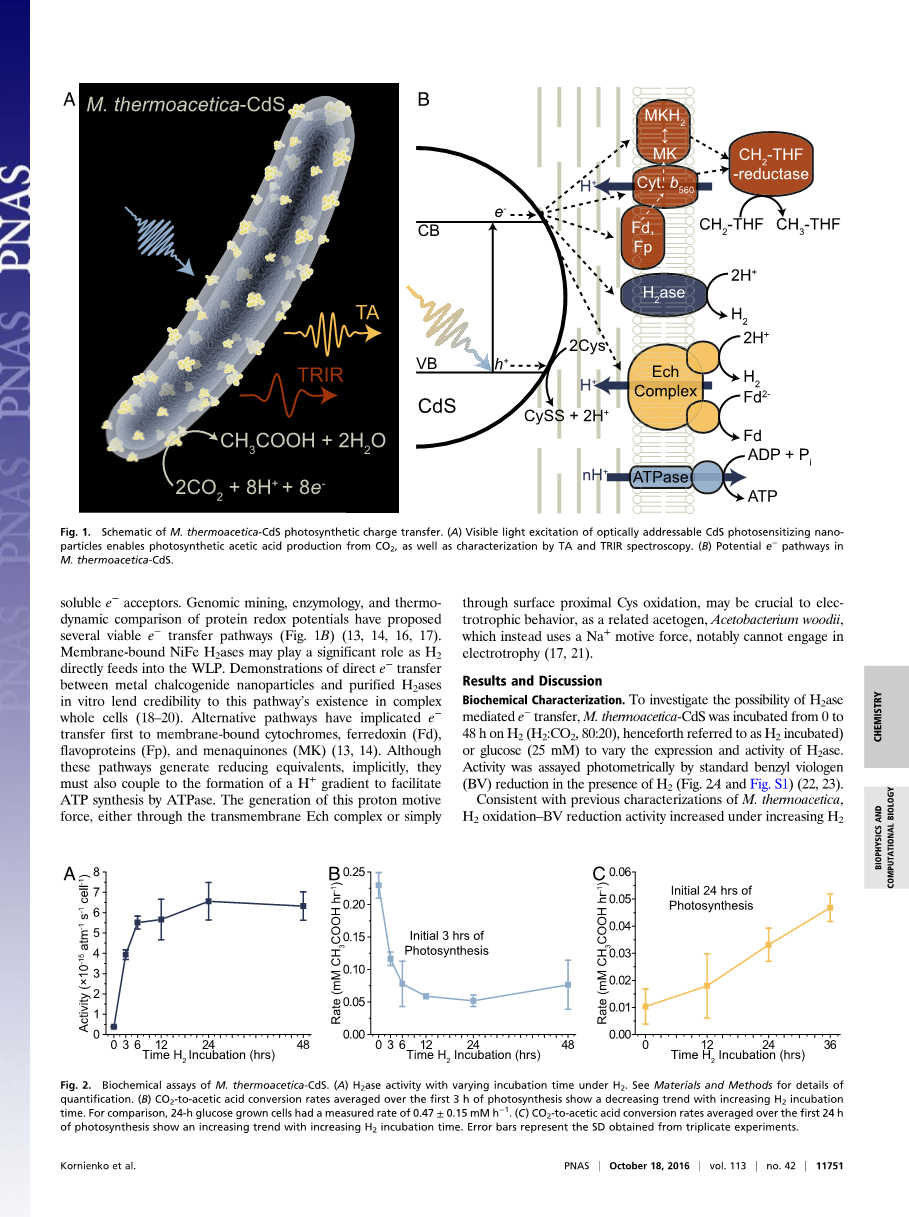
图1. M. thermoacetica-CdS光合作用电荷转移示意图(A)光学可寻址硫化镉光敏性纳米粒子的可见光激发能够利用二氧化碳进行光合乙酸生产,并通过TA和TRIR光谱进行表征.(B) M. thermoacetica-CdS中潜在的电子通路。
当CdS光激发时,半胱氨酸氧化为胱氨酸,并且H 使价带h 猝灭,而导带电子可能转移到膜结合蛋白或可溶性电子受体。蛋白质氧化还原电位的基因组采掘、酶学和热力学比较提出了几种可行的电子传输途径(图1B)(13,14,16,17)。膜结合镍铁氢化酶可能在氢气直接进入WLP时起重要作用。金属硫系化合物纳米粒子与体外纯化的氢化酶之间的直接电子转移证明了该途径在复杂的全细胞中的存在(18-20)。其他的途径首先涉及到电子到膜结合细胞色素、铁氧还蛋白、黄素蛋白和苯醌的转移(13,14)。尽管这些途径会产生还原当量,但也必须与H 梯度的形成相结合,以促进ATP酶合成ATP。这种质子动力的产生,无论是通过跨膜ech复合物还是通过表面近端半胱氨酸氧化,都可能是电营养行为的关键,因为作为一种相关的醋原,木醋酸杆菌,它使用了Na 动力,特别是不能参与电营养(17,21)。
结果和讨论
生化特性 为了研究氢化酶介导的电子转移的可能性,从0到48h将M. thermoacetica-CdS培养在氢气中(H2:CO2,80:20,所以称为H2培养)或者葡萄糖(25mM)中去改变氢化酶的表达和活性。在氢气存在的条件下用标准苄基紫精还原光度法测定活性(图2A和图S1) (22,23)。
图2 M. thermoacetica-CdS的生化分析。(A)在氢气存在的条件下不同培养时间氢化酶的活性,具体量化信息参见材料和方法。(B)在光合作用的前3小时内,CO2到乙酸的平均转化速率随H2培养时间的增加呈下降趋势。为了进行比较,24小时葡萄糖生长细胞的测量率为0.47plusmn;0.15 mM hminus;1。(C)在前24个小时光合作用中,平均的二氧化碳到乙酸的转化速率随H2培养时间的延长呈上升趋势。误差条代表从三次实验中得到的标准差。
与先前M. thermoacetica的特征一致,H2氧化-BV还原活性在增加H2培养时间下增加,可能是H2ase表达增加导致的。在葡萄糖和H2下培养24小时的M. thermoacetica-CdS比较显示,H2ase活性分别为6.56plusmn;0.92times;10-15(atm s cell)minus;1和1.85plusmn;8.39times;10-17(atm s cell)minus;1。
为了将酶活性与光合性能联系起来,对相同的样品进行太阳光照模拟(0.5%太阳光,AM1.5g)和乙酸生产分析。在光合作用的最初3小时内,二氧化碳减少率与氢化酶活性反相关,0小时(以前仅在葡萄糖上生长)培养样品显示出最高活性(0.23plusmn;0.02 mM h-1)。24小时葡萄糖培养样品以0.47plusmn;0.15 mM hminus;1的速率生产乙酸,比24 h H2培养样品(0.052plusmn;0.009 mmMHminus;1)快一个数量级。然而,在较长的光照时间,H2被培养样本显示出相反的趋势。当平均光照在24小时以上时,随着在氢气中培养时间的增加,光合反应速率从0小时氢气培养的0.010 plusmn; 0.001 mM hminus;1(CdS带隙以上光子的7.1plusmn;4.5%量子产额)增加到36小时氢气培养时的0.047 plusmn; 0.005 mM hminus;1(32plusmn;4%量子产额)(图2C)。我们注意到,前3小时内的醋酸生成率高于24小时的平均值。3-h的速率也不是化学计量的(例如,0-h H2样品的表观量子产额为160plusmn;10%),这表明尽管大量清洗和去除残留的葡萄糖和H2,一些还原的中间产物仍从预培养期携带过来。观察到的趋势表明,可能是通过在WLP中的一组较新的酶,较低H2ase活性的细胞更有效地转移硫化镉电子以最终减少这些中间产物。相比之下,24小时平均数据在量子产额上与之前的报告一致,没有发现这种残留中间产物(10)。这些对比结果表明两种相互竞争的电荷转移机制:一种在短时间内(lt;3小时)占主导地位的非氢化酶介导途径和在长时间内占主导地位的氢化酶介导的途径(~24小时)。
瞬态吸收光谱法。为深入研究这两种机制的分子基础,我们转向时间分辨光谱技术,动态了解活性趋势。瞬态吸收衰变动力学遵循光生电子离开CdS去往M. thermoacetica-CdS的多种准备的速率(图3和图S2A)。从440到490nm的瞬态漂白与用CdS 电子受体系统观察到的典型光谱相匹配,而在无CdS的M. thermoacetica中没有观察到。化学沉淀的CdS的衰减光谱比M. thermoacetica-CdS慢得多,表明快速淬灭可能是由近端电子受体引起的。氢气培养的与更高的氢化酶活性密切相关的M. thermoacetica-CdS与葡萄糖类似物相比,显示出更快的衰变动力学。这些观察结果表明,在H2培养的细菌中,可能更快地将电子传递到受体部位,或更多的电子受体(20)。将每一个瞬态吸收数据集拟合到三次衰变,显示出三种生命周期:2-10 ps(tau;1)范围内的快速分量,20-80 ps范围内的较长分量(tau;2),几百皮秒范围内的更长分量(tau;3)(表S1)。多指数衰减不出意料地表明,在复杂的M. thermoacetica-CdS混合物中有几个过程在起作用。先前用具有快速电子转移行为的胶态CdS的特征分子受体测量了快速皮秒衰变(24–26)。由于镉硫系量子点中的热电子弛豫发生在亚秒区,因此在测量的时间尺度中,这个过程不可能有助于瞬态吸收动力学(27)。然而,以前对FeFe H2ase-CdS结构的研究表明瞬态吸收的周期在100ns范围内,明显长于本文的数据(19,20)。这种差异可能是由于表面配体(不存在于M. thermoaceticaCdS中)存在电荷转移屏障、CdSH2as空间邻近性差异、溶剂效应和重组能、氢键网络以及纯化酶在体外和体内条件下的功能相对受损(28–30)。分子载体也可能发挥作用,接受电荷并随后将其转移到H2ase,以及其他潜在的电荷转移途径。
无细胞而只有CdS培养的细胞、H2培养细胞和葡萄糖培养细胞的寿命差异表明H2ase的表达在电子转移动力学中的重要性 。与上述氢化酶活性相关(图2a),测量了H2培养时间序列的瞬态吸收周期(图3b和c)。快速的tau;1、tau;2及其加权平均值均随H2ase活性的增加而寿命缩短,表明快速电子转移动力学是由于H2ase中电子受体位点的增加或性质与H2ase的表达相关的分子载体的增加。用CO抑制H2ase活性位点(H簇)并没有显著改变瞬态吸收动力学,与之前的研究类似,其中电子转移最初通过FeS簇链进行,而不是直接进入NiFe活性位点(图s2b)(20、31、32)。
时间分辨红外光谱法。尽管向膜结合的氢化酶的高效电子转移可能解释了长时间范围内光合作用速率的增加(图2c),但时间分辨红外法帮助确定了短时间范围内光合作用速率降低的基础(图2b)。
在氨基酸残基的其他红外有源模式特征中,我们观察到了1760–1880 cmminus;1光谱窗口中的变化,大致对应于CO和CN双键和三键的振动范围(图4)(33-35)。峰值与瞬态吸收信号在同一时间尺度上衰减(图S3–S6),进一步证明皮秒电子转移是由分子而非纯物理过程引
资料编号:[5634]
以上是毕业论文外文翻译,课题毕业论文、任务书、文献综述、开题报告、程序设计、图纸设计等资料可联系客服协助查找。
您可能感兴趣的文章
- 播撒生物炭促进鸟粪石形成,但加速重金属积累外文翻译资料
- 钢铁工业余热有机朗肯发电的能量及炯分析外文翻译资料
- 深度共晶溶剂微波辅助处理木质素-碳水化合物复合 物的高效裂解及超快提取木质素低聚物外文翻译资料
- 功能化杯状芳烃离子团族[4]的合成、晶体结构及竞争结合性能外文翻译资料
- 面向高能量密度柔性超级电容器的无纺布用黑磷杂化微纤维的微流控纺丝结构外文翻译资料
- 活性炭对水溶液中氨的吸附外文翻译资料
- 制备可控海胆状NiCo2S4微球协同硫掺杂石墨烯作为高性能 二次锌空气电池的双功能催化剂外文翻译资料
- 钛酸盐材料对重金属离子的吸附外文翻译资料
- CO2敏感催化剂的合成与表征温度响应催化聚离子液体微凝胶外文翻译资料
- 温度响应微凝胶薄膜在湿环境中作为可逆二氧化碳吸收剂外文翻译资料

