
英语原文共 8 页,剩余内容已隐藏,支付完成后下载完整资料
译文
基于聚合物供体和受体的二元共混物的全聚合物太阳能电池(all-PSCs)由于其相对于基于富勒烯的PSC的优势(例如增强的光吸收以及化学结构和能级的广泛可调性)而备受关注。此外,与基于富勒烯的PSC相比,all-PSCs具有增强的抵抗热应力和机械应力的稳定性,这对于生产灵活,便携式的PSC至关重要。最新的电源转换效率(PCE)all-PSCs的百分比均已超过8%,PCE高于7%的高效all-PSCs仍然非常罕见。总的来说,all-PSCs的PCE仍远低于使用相同的聚合物供体制备的基于富勒烯的PSC。all-PSCs的性能通常受到短路电流密度(Jsc)和填充因子(FF)的限制,这主要归因于(i)取向错误产生低电子迁移率的聚合物受体,(ii)供体/受体(D / A)界面处的电荷解离效率低,(iii)由于大力促进了聚合物与聚合物的混合,未优化的本体-异质结(BHJ)共混物形态,和(iv)相对聚合物受体的光吸收系数低。此外,尽管all-PSCs具有实现高开路电压(Voc)的巨大潜力,但由于聚合物受体的选择有限,要实现超过1 V的Voc值仍然具有挑战性。因此,设计具有互补的光吸收和合适的能级以同时最大化Jsc,FF和Voc值的合适的供体和受体聚合物对非常重要。 共轭聚合物的氟化已被证明是优化光学和电学性质的有效方法,从而增强了基于富勒烯的PSC的光伏性能。由于氟的鲍林电负性最高,为4.0,因此共轭聚合物的氟化可有效降低最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)能级,以控制电荷转移的能量偏移并提高PSC的Voc值。此外,在聚合物上引入氟原子可以在不引入空间位阻的情况下,通过C-F和H-C在聚合物中的强相互作用促进其分子平面性和分子间组装,从而提高了聚合物的电荷迁移率。此外,氟化可以有效地增强疏水性并调节聚合物的极性,从而控制界面相互作用其他聚合物或富勒烯,这通常会导致BHJ形态得到改善,具有较小的相域和较大的界面面积,从而增加激子解离并提 高Jsc和FF值。迄今为止,尽管其已广泛用于基于富勒烯的PSCs,但在all-PSCs系统中尚未对氟化进行深入研究。仁等。首次证明了氟化在all-PSCs中的有效性,其中基于氟化萘二酰亚胺(NDI)的聚合物受体在all-PSCs的形态和电荷传输性质方面产生了显着改善。然而,考虑到氟化对HOMO / LUMO水平,氟化策略应更好地用于聚合物供体,因为它可以降低其HOMO水平和增加Voc值。
本文中,我们开发了一系列新的聚合物供体(P1,P2和P3),并研究了氟化对all-PSCs中聚合物性能和光伏性能的影响。与之前讨论聚合物链主链氟化的大多数工作不同,我们在聚合物供体的侧链中引入了不同数量的氟原子(0、2和4),并系统地研究了侧链氟化对all-PSCs性能的影响。聚合物的侧链氟化对器件性能产生显着影响:PCE从基于P1的全PCS的2.93%分别增加到基于P3和P2的all-PSCs的5.21%和7.13%。首先,氟化的P2和P3聚合物的HOMO和LUMO含量逐渐降低,因此提高了all-PSCs的Voc值(P1器件为0.88 V,P2器件为0.94 V,对于P3器件为1.04 V)。另外,发现氟化是有利于通过抑制重组过程增强吸收系数和促进激子离解,从而显着提高Jsc和FF值。此外,聚合物的侧链氟化促进了具有良好混合的共混物结构域的良好BHJ形态。重要的是,氟化程度对all-PSCs的性能具有至关重要的影响。例如,由于增强的电性能,即高电荷迁移率、平衡的mu;h/mu;e比和更好的电荷解离,P2器件(两个氟原子,7.13%)而不是P3器件(四个氟原子,5.21%)获得了最佳效率。因此,我们的侧链氟化提供了优化all-PSCs中聚合物的结构,电化学,电和光伏性质的优势。
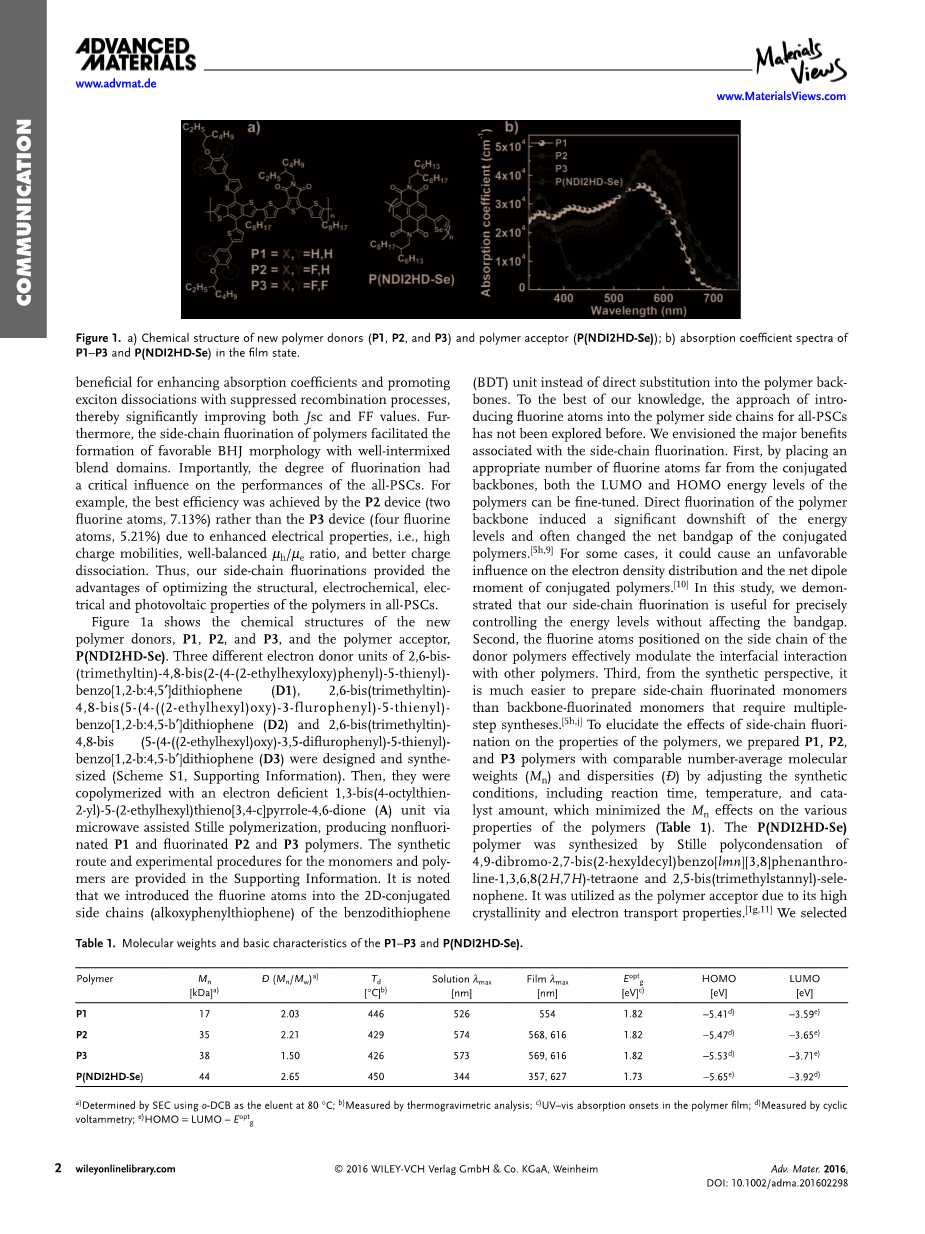
图1a显示了新的聚合物供体P1,P2和P3,以及聚合物受体P(NDI2HD-Se)的化学结构。2,6-双(三甲基锡)-4,8-双(2-(4-(2-(2-乙基己氧基氧基)苯基)-5-噻吩基)-苯并[1,2-b:4,5]的三个不同的电子供体单元 ] dithiophene(D1),2,6-双(三甲基锡)-4,8-(5-(4-(((2-乙基己基)氧基)-3-氟苯基)-5-噻吩基)-苯并[1, 2-b:4,5-b ]二噻吩(D2)和2,6-双(三甲基锡)-4,8-双(5-(4-(((2-乙基己基)氧基)-3,5-二氟苯基)设计并合成了)-5-噻吩基)-苯并[1,2-b:4,5-b ]二噻吩(D3)。然后,将它们与缺电子的1,3-双(4-辛基噻吩-2-基)-5-(2-乙基己基)噻吩并[3,4-c]吡咯-4,6-二酮(A)单元共聚协助Stille聚合,生产非氟化P1以及氟化P2和P3聚合物。支持信息中提供了单体和聚合物的合成路线和实验步骤。注意,我们将氟原子引入了苯并二噻吩的2D共轭侧链(烷氧基苯基噻吩)中(BDT)单元,而不是直接取代到聚合物主链中。据我们所知,以前从未探索过将氟原子引入all-PSCs的聚合物侧链的方法。我们设想了与侧链氟化相关的主要优点。首先,通过将适当数量的氟原子置于远离共轭骨架的位置,可以对聚合物的LUMO和HOMO能级进行微调。聚合物主链的直接氟化作用引起能级的显着下降,并经常改变共轭聚合物的净带隙。在某些情况下,它可能对共轭电子的电子密度分布和净偶极矩产生不利影响聚合物。在这项研究中,我们证明了我们的侧链氟化对于精确控制能级而不影响带隙非常有用。其次,位于供体聚合物侧链上的氟原子有效地调节了与其他聚合物的界面相互作用。第三,从合成的角度来看,制备侧链氟化单体比需要多步合成的骨架氟化单体要容易得多。为了阐明侧链氟化对聚合物性能的影响,我们制备了P1 ,P2和P3聚合物,具有相当的数均分子量(Mn)和分散度(Đ),可通过调节合成条件(包括反应时间,温度和催化剂量)来最大程度地降低Mn对聚合物的影响聚合物的各种特性(表1)。通过4,9-二溴-2,7-双(2-己基癸基)苯并[lmn] [3,8]菲咯啉-1,3,6,8(2H)的Stille缩合反应合成P(NDI2HD-Se)聚合物,7H)-四酮和2,5-双(三甲基锡烷基)-硒烯。由于其高的结晶度和电子传输性能,它被用作聚合物受体。我们为NDI单元选择了2-己基癸基(HD)侧链,以促进具有表面几何形状的晶体堆积。
表1总结了聚合物的分子量和基本特性。图1b和图S1比较了它们在薄膜状态和溶液中的UV-可见吸收光谱。由吸收开始产生的P1,P2和P3的光学带隙等于1.82eV。在554 nm处测量了P1膜的最大吸收(lambda;max),而氟化P2和P3膜的吸收光谱在568 nm处显示了类似的红移 最大值,在616 nm处有很强的振动肩峰。表明侧链氟化改善了分子间的相互作用。重要的是, 最大值处的吸收系数也从3.6times;104cm 1(对于P1)增加到4.8times;104cm 1(对于P2)和4.5times;104 cm 1(用于P3),证明了氟化对提高光收集能力的有益作用。
如图S2所示,通过循环伏安(CV)测量来检查每种聚合物的能级。氟化有效地降低了HOMO的水平,这是因为氟原子具有很强的吸电子能力,即-5.41(P1),-5.47(P2)和-5.53 eV (P3).此外,LUMO的能级降低了从P1聚合物的-3.59eV到P2和P3聚合物的-3.65和-3.71eV。假设测得的P(NDI2HD-Se)聚合物受体的LUMO和HOMO含量分别为-3.92和-5.65eV,则P1,P2和P3聚合物具有足够的LUMO / HOMO偏移量,以实现有效的激子离解。 D / A聚合物。为了确认氟化对能级的这种影响,使用密度泛函理论(DFT)计算了P1,P2和P3聚合物的分子几何形状和电子密度分布。DFT计算使用高斯09程序包中设置的B3LYP函数和6–31G(d)为基础进行。P1-P3的单个重复单元用于简化计算并阐明氟化在分子能级上的作用。随着所添加氟原子数量的增加,聚合物主链上的轨道变得更加离域,从而逐渐降低HOMO / LUMO能级。氟化的P2和P3低于P1,即-4.98(P1),-5.07(P2)和-5.17eV(P3)。有趣的是,尽管侧链氟化有效地降低了P2和P3聚合物的HOMO和LUMO含量,并显着提高了光吸收系数,但所有三种聚合物的光学带隙均相同。这凸显了我们进行侧链氟化的分子设计的重要性,这与通常引起带隙变化的其他骨架氟化不同。
氟化作用对铝的光电性能的影响 通过评估基于P1:P(NDI2HD-Se)-,P2:P(NDI2HD-Se)-和P3:P(NDI2HD-Se)的all-PSCs,探索了all-PSCs。BHJall-PSCs器件采用ITO / ZnO /聚合物共混物/ MoO3/ Ag的倒置结构制造。对于三种不同的all-PSCs,D:A的混合重量比和有源层的厚度分别优化为1:1和 100nm。所有聚合物共混物均由氯仿与2体积%的二苯醚(DPE)制成,所得共混物层用1-丁醇处理。图2c,d显示了all-PSCs的代表性电流密度-电压曲线(J–V)和外部量子效率(EQE)。表2列出了器件的特性。如表2所示,氟化聚合物供体(P2和P3)的使用显着增加了所有光伏电池的性能。all-PSCs中的参数(Jsc,Voc,和FF)。由于HOMO能级的逐步下降,基于P2和P3的all-PSCs表现出增强的Voc值(P2为0.94 V,P3为1.04 V,而P1为0.88 V)。在基于P2和P3的all-PSCs中,Jsc和FF值也得到了改善。因此,对于P1:P(NDI2HD-),all-PSCs的所得PCE值从2.93%(Voc:0.88 V,Jsc:7.41 mA·cm 2,FF:0.45)显着增加。 Se)至5.21%(Voc:1.04 V,P3:P(NDI2HD-Se)的Jsc:9.27 mA·cm 2和FF:0.54),达到7.13%(Voc:0.94 V,Jsc:12.22 mA·cm 2和FF:0.62)P2:P(NDI2HD-Se).有趣的是,器件性能在很大程度上取决于侧链氟化的程度。由于提高了Jsc和FF值,P2设备在较低的氟化度下获得了最佳效率。如图2d中的EQE曲线所示,器件的Jsc值的差异很好地反映在其光谱响应的变化中。所有的Jsc值都与EQE谱计算的Jsc值完全匹配(误差在3%以内)。如图2d所示,在整个可见范围内,基于P2的all-PSCs的EQE值均高于基于P1和基于P3的all-PSCs的EQE值,令人印象深刻的EQE值为75.4%。基于P2的all-PSCs的高Jsc值部分归因于其共混膜的光吸收增强,P2的最大吸收系数为4.2times;104cm 1 :P(NDI2HD-Se)胶片相反,P1:P(NDI2HD-Se)膜和P3:P(NDI2HD-Se)膜的值为分别为3.3times;104cm 1和3.9times;104cm 1(图S4,支持信息)。然而,共混膜的不同光吸收值可能不是Jsc值显着提高的唯一原因,即与P1相比,P2增强了65%。因此,我们检查了all-PSCs的电性能,包括电荷传输和激子离解几率。
首先,我们研究了P1:P(NDI2HD-Se),P2:P(NDI2HDSe)和P3:P(NDI2HD-Se)的空穴迁移率(mu;h)和电子迁移率(mu; e) -聚合物膜,使用空间电荷限制电流(SCLC)方法在最佳设备条件下制备。表3和图S5显示了测得的mu; h和mu; e值如表3所示,P2:P(NDI2HD-s e)和P3:P(NDI2HD-Se)薄膜具有高mu;e值(分别为7.73times;10-5和5.38times;10-5 cm2 V-1s-1),比P1:P(NDI2HD-Se)混合物的(4.36times;10-6cm2v-1s-1)高一个数量级,这表明P2和P3共混膜中从活性层到电极的电荷通道形成得更好。并且,P2:P(NDI2HD-Se)膜的 h值最高,为1.12times;10 4cm2V1s1,它比P1:P(NDI2HD-Se)膜增加了两倍(5.62times;10 5cm2V1s1),比P3:P(NDI2HD-Se)膜增加了十倍( 1.31 10 5cm2V1s1)。聚合物的较高氟化度(P3)降低mu; h,因为聚合物的过量氟化通常会降低其溶解度并引起溶液中的强聚集,从而影响聚合物的堆积结构和薄膜取向。因此,与P1共混膜(mu;h/mu;e=12.89)和P3共混膜(mu;h/mu;e=0.24)相比,P2共混膜(mu;h/mu;e=1.45)的mu;h/mu;e比率更为平衡。这种特性对于减少空间电荷的积累,从而提高P2共混膜的FF值是非常有益的。
为 了了解每 种共混物 的电荷重 组行为, 我们针对 P1:P(NDI2HD-Se),P2:P(NDI2HD-Se)和P3:P(NDI2HD-Se)胶片。图S6和图3a显示了Jsc的对数-对数图和Voc的对数-线性图,它是来自0.1到1个太阳,这说明了器件中重组过程的类型。通常,Jsc与P的幂律相关性(Jsc P )和 值当由于双分子重组引起的光电流损失可忽略不计时,趋近于1。如图S6所示,P2共混物的 值(0.99)比P1共混物(0.96)和P3共混物(0.97)更接近于1。尽管差异不明显,但结果表明,在基于P2的all-PSCs中,双分子重组被最有效地抑制。根据P值,差异在Voc趋势中更清楚地显示(图3a)。Voc与ln(P)之间的关系中的斜率(S)的大小可以作为区分主要重组机制的指标。P1:P(NDI2HD)的S值从1.32显着降低-Se)all-PSC对于P2:P(NDI2HD-Se)al-PSCs为1.06。而且,P2:P(NDI2HD-Se)all-PSC的该值显着低于P3:P(NDI2HD-Se)all-PSC的(1.20),这表明单分子和/或陷阱辅助的重组大大降低了。由于有效的空穴/电子传输以及D / A界面上的电荷转移,P2:P(NDI2HD-Se)all-PSCs。
接下来,我们通过绘制光电流密度(Jph值)曲线与有效电压(V效)的曲线图,比较了三种all-PSCs共混物的激子解离几率(P(E,T))。根据确定的程序,我们测量了从-10到2V的宽电压(Va)的Jph曲线,并使用公式V效计算了Veff=V0 -Va,其中V0是Jph值 0时的电压。每个器件的Jph值当V效接近10 V时变得饱和以产生饱和光电流密度(J坐着),并使用等式(J
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[410057],资料为PDF文档或Word文档,PDF文档可免费转换为Word

